Using Folded Proteins as Mechanically Well-Defined Units to Understand Fatigue Fracture in Hydrogels: Bridging Single Molecule and Bulk Studies
ISOM keeps this Physical Review X paper in the public review set because it gives readers a concrete case around Using Folded Proteins as Mechanically Well-Defined Units to Understand Fatigue Fracture in.
Background & Academic Lineage
The Origin & Academic Lineage
Hydrogels are widely used in applications requiring durability under cyclic loading, such as artificial cartilage and soft robotics. However, their reliability is often limited by fatigue fracture, which involves the gradual growth of cracks under repeated subcritical loads. This problem has been a persistent challenge in materials science, particularly in reconciling experimental observations of hydrogel fracture with classical theoretical modles.
Historically, the Lake-Thomas theory [6] provided an initial framework, estimating fracture energy based on the rupture of a single layer of polymer chains. However, this approach fundamentally underestimated the energy dissipation observed during actual crack propagation [7]. This significant discrepancy highlighted a critical "pain point": real hydrogel networks dissipate far more energy than simple theories predicted, necessitating improved theoretical frameworks. Subsequent efforts, like those by Zhao and co-workers, introduced nonlocal dissipation concepts [7,8], where bond breaking extends into a broader region of the network, partially bridging the gap between theory and experiment. Craig and colleagues further refined these models by incorporating nonlinear elasticity of polymer strands [9]. Despite these advancements, a fully quantitative agreement between theory and experiment remained elusive, primarily due to the complex, multiscale nature of bond breaking in tough hydrogels.
Another line of research explored mechanophores—force-sensitive covalent crosslinkers—as tools to probe and control hydrogel mechanisms [11]. While promising, mechanophores faced their own inherent limitations: their activation involves irreversible bond rupture, which depletes network integrity over time [13-16]. Furthermore, if the activation force of mechanophores was too high, non-specific backbone scission could occur, complicating the direct correlation between crosslinker strength and fracture energy [11,17]. These challenges motivated the search for alternative strategies that could offer reversible energy dissipation without compromising network integrity. This paper directly addresses these limitations by introducing folded protein domains as reversible, mechanically defined sacrificial units, aiming to bridge the gap between molecular-scale events and bulk fatigue behavoir.
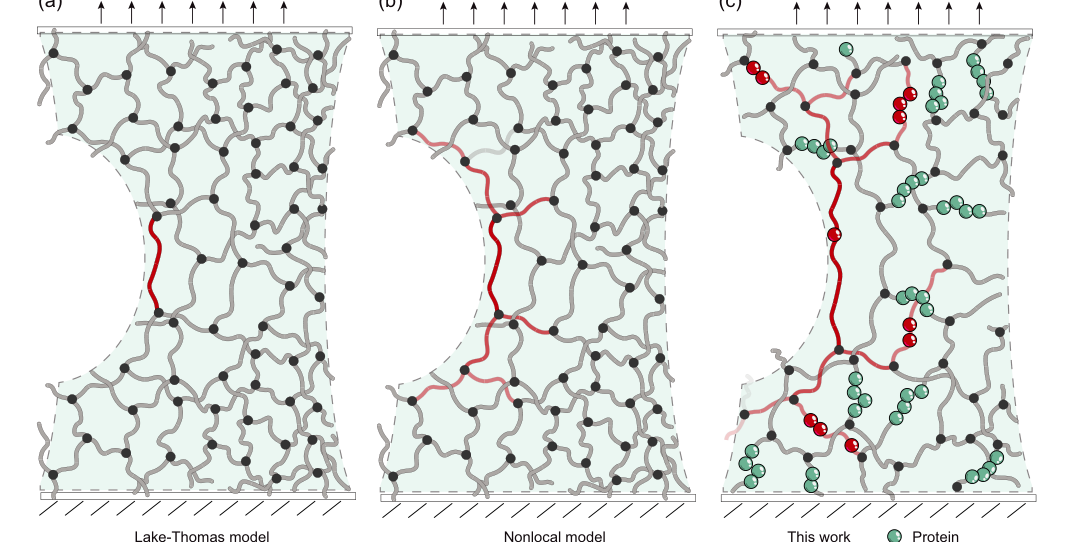 FIG. 1. Conceptual scheme of energy dissipation during crack propagation. (a) In the Lake-Thomas model, the intrinsic fracture energy equals the energy needed to break a single layer of polymer chains. (b) The intrinsic fracture energy mainly results from nonlocal energy dissipation by relaxing polymer chains far away from the crack tip. (c) In our force-response model, we suggest that force transduction at the crack tip through the polymer network couples with the conformational change of the polymer leading to an expanded crack zone. In our case the conformational change of polymer involves force-dependent protein unfolding or folding
FIG. 1. Conceptual scheme of energy dissipation during crack propagation. (a) In the Lake-Thomas model, the intrinsic fracture energy equals the energy needed to break a single layer of polymer chains. (b) The intrinsic fracture energy mainly results from nonlocal energy dissipation by relaxing polymer chains far away from the crack tip. (c) In our force-response model, we suggest that force transduction at the crack tip through the polymer network couples with the conformational change of the polymer leading to an expanded crack zone. In our case the conformational change of polymer involves force-dependent protein unfolding or folding
Intuitive Domain Terms
Here are some specialized terms from the paper, explained with everyday analogies:
- Hydrogels: Imagine a super-absorbent jelly or a contact lens. Hydrogels are soft, water-filled materials, mostly liquid but held together by a network of polymer chains, giving them a solid-like shape.
- Fatigue Fracture: Think about bending a paperclip back and forth repeatedly. Even if you don't bend it hard enough to break it in one go, it will eventually snap after many cycles. Fatigue fracture in materials is a similar process: gradual damage and crack growth under repeated, sub-critical stresses.
- Mechanophores: These are like tiny, one-time-use "stress sensors" or "sacrificial links" built into a material. When a certain force is applied, they break or change color, signaling that the material is under strain. The catch is, once they've done their job, they're gone for goood.
- Folded Protein Domains: Picture a miniature, biological spring or a tiny, reusable shock absorber. These are specific sections of proteins that can unfold (absorb energy) when pulled with enough force and then refold (recover) when the force is released, without permanently breaking the material. They are the key innovation in this paper.
- Energy Release Rate ($\Gamma$): This is like the "fuel" or "driving force" for a crack to grow in a material. A higher energy release rate means there's more energy available to extend the crack, making the material more prone to fracture.
Notation Table
| Notation | Description | Type |
|---|---|---|
| $F_i$ | Force in the $i$-th layer of the polymer network | Variable |
| $s$ | Singular exponent reflecting spatial decay of force distributions | Parameter |
| $k_{uf,i}$ | Unfolding rate of protein domains in the $i$-th layer | Variable |
| $k_{f,i}$ | Refolding rate of protein domains in the $i$-th layer | Variable |
| $z_{uf}$ | Unfolding transition distance of protein domains | Parameter |
| $z_f$ | Refolding transition distance of protein domains | Parameter |
| $k$ | Boltzmann constant | Parameter |
| $T$ | Absolute temperature | Parameter |
| $\Gamma$ | Fatigue threshold (maximum energy release rate before crack propagation) | Variable |
| $U$ | Total energy consumed within the polymer network | Variable |
| $N_{chain}$ | Chain density of fabricated hydrogels | Parameter |
| $L_0$ | Initial length of the polymer chain | Parameter |
Problem Definition & Constraints
Core Problem Formulation & The Dilemma
The core problem this paper addresses is the persistent challenge of understanding and enhancing the fatigue resistance of hydrogels, particularly under cyclic loading.
Input/Current State: Hydrogels are widely utilized in diverse applications, from artificial cartilage to soft robotics, where durability under repeated mechanical stress is paramount. However, their reliability is severely limited by their notorious susceptibility to fatigue fracture. The current understanding of hydrogel fatigue is incomplete, largely due to the intricate, multiscale interplay between molecular-level events (like bond breaking) and macroscopic crack propagation. Existing theoretical models, such as the Lake-Thomas theory, significantly underestimate the actual energy dissapation observed during crack propagation, indicating a fundamental gap in our understanding of how these materials dissipate energy. Furthermore, previous strategies employing mechanophores for energy dissipation relied on irreversible bond rupture, which inherently compromises the long-term integrity of the network by depleting sacrificial units over time. These prior models also often oversimplified the network structure, assuming homogeneous chain behavior and uniform force decay.
Desired Endpoint/Goal State: The ultimate goal is to develop next-generation, fatigue-resistant hydrogels that not only exhibit enhanced durability but also retain low stiffness and high extensibility. This requires a comprehensive, quantitative framework that can accurately predict and explain hydrogel fatigue behavior by directly correlating molecular-scale events with macroscopic material properties. Specifically, the authors aim to overcome the limitation of irreversible energy dissipation by introducing reversible sacrificial units and to establish a model that precisely couples force redistribution within the network with the kinetic unfolding and refolding of these molecular domains. This framework should enable a rational design principle for hydrogels, moving beyond trial-and-error.
Exact Missing Link or Mathematical Gap: The precise missing link is a quantitative, multiscale model that bridges the gap between the force-induced conformational changes of individual polymer segments (specifically, protein unfolding/refolding) and the macroscopic propagation of fatigue cracks. Previous models failed to accurately capture the complex, non-uniform energy dissipation mechanisms. This paper attempts to bridge this gap by:
1. Introducing a singular force decay law across network layers, given by $F_i = F_1 \cdot i^s$ and $m_i = i^{-s}$ (Equation 1), which aligns with crack-tip stress singularity principles and accounts for the spatial decay of force distributions more realistically than previous exponential or uniform decay assumptions.
2. Incorporating the force-response unfolding and refolding kinetics of folded protein domains based on Bell's theory, where the unfolding and refolding rates are described by:
$$k_{uf,i} = k_{uf}^0 \exp\left(\frac{F_i z_{uf}}{kT}\right) \quad \text{and} \quad k_{f,i} = k_f^0 \exp\left(-\frac{F_i z_f}{kT}\right)$$
(Equation 2), which explicitly accounts for the time and cycle-dependent nature of energy dissipation and the probabilistic failure/reformation of bonds. This mathematical integration allows for a more realistic cascade of multistep kinetic events, rather than assuming uniform or instantaneous bond breaking.
The Dilemma: The most painfull trade-off that has trapped previous researchers is the inherent conflict between enhancing fatigue resistance and preserving other critical material properties or network integrity.
1. Fatigue Resistance vs. Network Integrity: Traditional approaches to improve fatigue resistance often involve sacrificial bonds that irreversibly break to dissipate energy. While this provides immediate toughening, it leads to a gradual depletion of the network's load-bearing capacity and overall integrity over repeated cycles, ultimately limiting the material's long-term durability.
2. Fatigue Resistance vs. Stiffness/Extensibility: Many strategies to increase hydrogel toughness and fatigue resistance, such as hierarchical designs or dynamic bonds, frequently result in an undesirable increase in material stiffness [36,42]. This is a significant drawback for applications requiring soft, highly extensible materials, creating a dilemma where improving one aspect (fatigue resistance) compromises another (softness and stretchability).
Constraints & Failure Modes
The problem of understanding and designing fatigue-resistant hydrogels is insanely difficult due to several harsh, realistic constraints:
- Multiscale Complexity: The fundamental challenge lies in the multiscale nature of the problem, where macroscopic fatigue crack propagation is governed by molecular-scale events (e.g., protein unfolding, bond rupture, chain relaxation). Reconciling these vastly different length and time scales in a coherent theoretical and experimental framework is extremely challenging.
- Irreversible Damage Accumulation: In many materials, damage accumulates irreversibly, leading to a progressive loss of structural integrity. For hydrogels, previous mechanophore-based designs suffered from this, as their activation involved permanent bond rupture, which depletes the network over time [13-16].
- Non-uniform Stress Distribution: Under loading, especially near crack tips, stress is highly concentrated and non-uniformly distributed across the polymer network. Accurately modeling how forces are transduced and dissipated through such heterogeneous networks is complex, as simplified assumptions of uniform force decay or homogeneous chain behavior are insufficient.
- Kinetic and History-Dependent Behavior: The energy dissipation mechanisms, particularly those involving protein unfolding and refolding, are not instantaneous but are kinetic and history-dependent. This means the material's response is influenced by the loading rate, frequency, and prior deformation history, making it difficult to predict behavior under dynamic cyclic loading conditions.
- Non-monotonic Dependence on Molecular Parameters: The paper reveals a non-monotonic relationship between fatigue resistance and the unfolding force of sacrificial domains. There is an optimal intermediate force window; if the unfolding force is too low, domains are prematurely exhausted, and if too high, they remain inactive. This complex dependence means simple monotonic optimization strategies will fail.
- Experimental Challenges in Correlation: Directly correlating single-molecule force spectroscopy experiements (AFM-SMFS) with bulk material fatigue tests requires meticulous control over molecular design (e.g., tunable unfolding forces, number of domains per crosslink) and precise macroscopic mechanical testing conditions. Ensuring that the molecular-level observations are truly representative of the bulk material's behavior is a significant experimental hurdle.
- Hardware Memory Limits (Implied): While not explicitly stated as a hard limit, the complexity of simulating large polymer networks with detailed kinetic models for each molecular unit implies significant computational demands. The use of "coarse-grained polymer-network simulation" as an independent validation suggests that more detailed, atomistic simulations might be computationally prohibitive for the entire system.
Why This Approach
The Inevitability of the Choice
The adoption of folded protein domains as reversible sacrificial units, coupled with a novel force-response model, was not merely an incremental improvement but a necessary paradigm shift. The authors realized that traditional "state-of-the-art" (SOTA) methods and existing theoretical frameworks were fundamentally insufficient to achieve a comprehensive understanding and practical design of fatigue-resistant hydrogels.
Specifically, the Lake-Thomas theory [6], a classical model for fracture energy, consistently "significantly underestimate[d] the energy dissapation observed during crack propagation [7]." While subsequent advances, such as nonlocal dissipation concepts [7,8] and modifications by Craig and colleagues [9] incorporating nonlinear elasticity, partially closed this gap, "fully quantitative agreement between theory and experiment remain[ed] elusive, largely due to the multiscale nature of bond breaking in tough hydrogels." These prior models often "oversimplified the system by assuming homogeneous chain behavior and uniformly folded domains," failing to capture the intricate, multiscale interplay between molecular events and macroscopic crack propagation.
Furthermore, the prevailing approach using small-molecule mechanophores, while innovative, faced inherent limitations. Mechanophores signal energy dissipation through "irreversible bond rupture," which "can deplete network integrity over time [13-16]." This one-time activation mechanism meant that the network's ability to dissipate energy diminished with repeated loading, ultimately compromising long-term durability. Moreover, issues like "nonspecific backbone scission" could occur if activation forces were too high, complicating the direct correlation between crosslinker strength and fracture energy [11,17]. These critical shortcomings in both theoretical modeling and material design necessitated an alternative strategy that could offer reversible energy dissipation and a more accurate, multiscale theoretical framework.
Comparative Superiority
This approach offers qualitative superiority over previous methods by fundamentally altering the mechanism of energy dissipation and providing a more robust theoretical framework.
- Reversible Energy Dissipation and Network Integrity: Unlike small-molecule mechanophores that rely on irreversible bond rupture, folded protein domains act as "reversable, mechanically defined sacrificial units." When subjected to stress, these domains unfold, dissipating energy, but crucially, "unfolding does not break the polymer backbone; rather, it extends the protein segment while preserving network connectivity." This allows the network to maintain its structural integrity and dissipate energy repeatedly over many cycles, a critical advantage for fatigue resistance under cyclic loading.
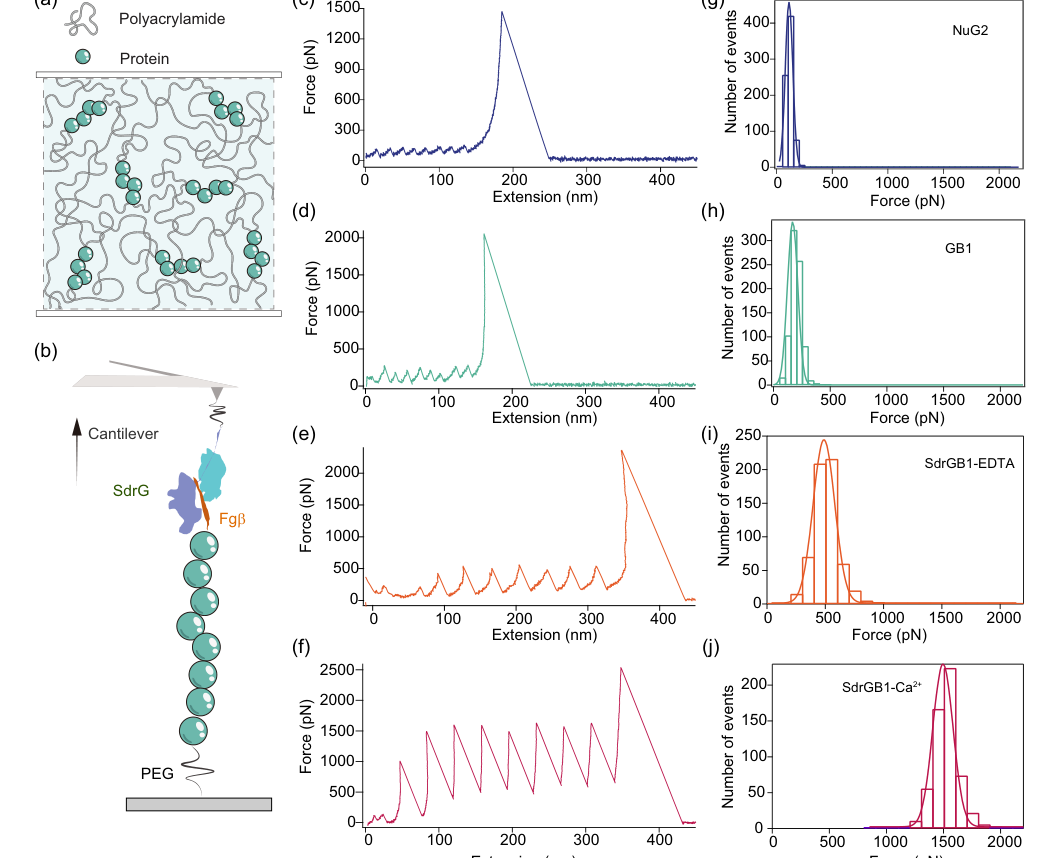 FIG. 2. Design principle for the antifatigue hydrogels and molecular mechanics characterization. (a),Schematic illustration of the hydrogel, in which the polyprotein as crosslinkers and polyacrylamide as the percolating phase to form random polymer network. (b) Schematic of the AFM based single molecule force spectroscopy experiments. (c)–(f) Representative single molecule force- extension curves of ðNuG2Þ8, ðGB1Þ8, and ðSdrGB1Þ8 with EDTA and Ca2þ, respectively. (g)–(j) The histograms of unfolding force of NuG2, GB1, SdrGB1-EDTA, and SdrGB1-Ca2þ, respectively. By fitting with Gaussian fit, the unfolding forces are 112.6 47.2 pN (mean S:D:) for NuG2 (n ¼ 758),193.3 64.8 pN for GB1 (n ¼ 794),474.6 131.6 pN for SdrGB1 with EDTA (n ¼ 601), and 1492.7 130.5 pN for SdrGB1 with Ca2þ (n ¼ 565), respectively. n is the total number of events
FIG. 2. Design principle for the antifatigue hydrogels and molecular mechanics characterization. (a),Schematic illustration of the hydrogel, in which the polyprotein as crosslinkers and polyacrylamide as the percolating phase to form random polymer network. (b) Schematic of the AFM based single molecule force spectroscopy experiments. (c)–(f) Representative single molecule force- extension curves of ðNuG2Þ8, ðGB1Þ8, and ðSdrGB1Þ8 with EDTA and Ca2þ, respectively. (g)–(j) The histograms of unfolding force of NuG2, GB1, SdrGB1-EDTA, and SdrGB1-Ca2þ, respectively. By fitting with Gaussian fit, the unfolding forces are 112.6 47.2 pN (mean S:D:) for NuG2 (n ¼ 758),193.3 64.8 pN for GB1 (n ¼ 794),474.6 131.6 pN for SdrGB1 with EDTA (n ¼ 601), and 1492.7 130.5 pN for SdrGB1 with Ca2þ (n ¼ 565), respectively. n is the total number of events
- Dynamic Stress Redistribution without Stiffness Compromise: The new network architecture introduces "spatially and temporally localized energy dissipation through force-activated protein unfolding." This enables the network to "dynamically redistribute stress under nonuniform deformation while preserving its bulk elastic integrity." Importantly, this enhancement in fatigue resistance is achieved "without increasing stiffness," which is a significant advantage over other toughening strategies like hierarchical designs or dynamic bonds that often lead to undesirable increases in stiffness [36,42].
- Advanced Multiscale Modeling: The developed force-response model is qualitatively superior because it "directly couples force redistribution with the kinetic unfolding or refolding of folded domains, resolving limitations in existing frameworks." It explicitly integrates Bell's law [28] for unfolding and refolding kinetics, allowing for "residual unfolded fractions to persist across cycles." This treatment captures the "intrinsically nonequilibrium and history-dependent nature of the dissipation process," providing a far more realistic representation of fatigue behavior than models assuming homogeneous chain behavior or uniform domain responses.
- Tunable and Predictive Design Principles: The use of engineered protein domains with "tunable unfolding forces (100–1500 pN)" and variable numbers of domains per crosslink (1 to 16) provides unprecedented control over material properties. The model reveals a "nonmonotonic dependence of fatigue resistance on the unfolding force," establishing an "optimal performance... within an intermediate force window." This allows for a "rational design principle" where unfolding force, refolding kinetics, and network connectivity can be jointly tuned for predictive rather than trial-and-error material design.
Alignment with Constraints
The chosen method—incorporating folded protein domains as reversible sacrificial units within polyprotein crosslinkers and developing a force-response model—perfectly aligns with the inherent constraints of understanding and designing fatigue-resistant hydrogels.
- Addressing Multiscale Complexity: A primary constraint was the "complex interplay between molecular-scale events and macroscopic crack propagation" that made quantitative agreement elusive. The force-response model directly addresses this by "coupling force-induced conformational change of polymer at the molecular level to the crack propagation at the macroscopic level [Fig. 1(c)]." It explicitly integrates molecular unfolding/refolding kinetics into a macroscopic fracture model, bridging the scales.
- Durability under Cyclic Loading: Hydrogels require "durability under cyclic loading," but fatigue fracture limits reliability. The "reversible" nature of folded protein unfolding, which dissipates energy without breaking the polymer backbone, is a direct solution. This allows the hydrogel to sustain repeated loads by continuously dissipating energy and "healing" at the molecular scale, thereby enhancing fatigue resistance.
- Maintaining Low Stiffness and High Extensibility: A crucial requirement for many hydrogel applications is to "retain low stiffness and high extensibility." Unlike other toughening strategies that often increase stiffness, this approach "enhances the fatigue threshold without compromising softness," offering a pathway to design fatigue-resistant hydrogels that maintain their desirable mechanical properties.
- Tunable and Programmable Properties: The ability to engineer protein domains with "tunable unfolding forces" and vary the "number of domains per crosslink" directly meets the need for programmable control over material behavior. This allows for systematic investigation and optimization of fatigue resistance based on molecular parameters.
- Quantitative Predictive Framework: The constraint of reconciling experimental observations with theoretical predictions, particularly the elusive "fully quantitative agreement," is met by the refined force-response model. The model's predictions "better match experimental observations, particulary in capturing the influence of domain unfolding or refolding kinetics and the number of domains on fatigue thresholds [Fig. 5(b)]," providing a robust quantitative framework.
Rejection of Alternatives
The paper explicitly or implicitly rejects several alternative approaches based on their fundamental limitations in addressing the problem of hydrogel fatigue.
- Traditional Fracture Models (e.g., Lake-Thomas theory [6]): These models were rejected because they "significantly underestimate the energy dissipation observed during crack propagation [7]." They failed to account for the complex, nonlocal energy dissipation mechanisms in real hydrogel networks.
- Advanced Nonlocal Models (e.g., Zhao and co-workers [7,8], Craig and colleagues [9]): While these models improved upon Lake-Thomas theory by introducing nonlocal dissipation and nonlinear elasticity, they were still deemed insufficient. The authors noted that "fully quantitative agreement between theory and experiment remain[ed] elusive, largely due to the multiscale nature of bond breaking in tough hydrogels." Furthermore, these "prior nonlocal models with hierarchical treelike networks oversimplified the system by assuming homogeneous chain behavior and uniformly folded domains," failing to capture the kinetic and history-dependent nature of molecular events.
- Small-Molecule Mechanophores: This popular approach was rejected due to its inherent constraints. Mechanophores rely on "irreversible bond rupture" for energy dissipation, which "can deplete network integrity over time [13-16]." This one-time activation mechanism is unsuitable for materials requiring long-term durability under cyclic loading. Additionally, issues like "nonspecific backbone scission" could occur if activation forces were too high, complicating the correlation between crosslinker strength and fracture energy [11,17].
- Other Toughening Strategies (e.g., hierarchical designs [36–38], dynamic bonds [39–41]): While these methods can "improve fatigue resistance," they were implicitly rejected for this specific application because "they often increase stiffness [36,42]," which is "undesirable for applications requiring high extensibility and low stiffness." The goal was to enhance fatigue resistance without compromising the hydrogel's inherent softness and flexibility.
Mathematical & Logical Mechanism
The Master Equation
The core of this paper's "force-response model" is built upon two fundamental sets of equations that describe how mechanical forces are distributed within the hydrogel network and how individual protein domains respond kinetically to these forces. These mechanisms collectively dictate the energy dissipation and, ultimately, the fatigue resistance of the material.
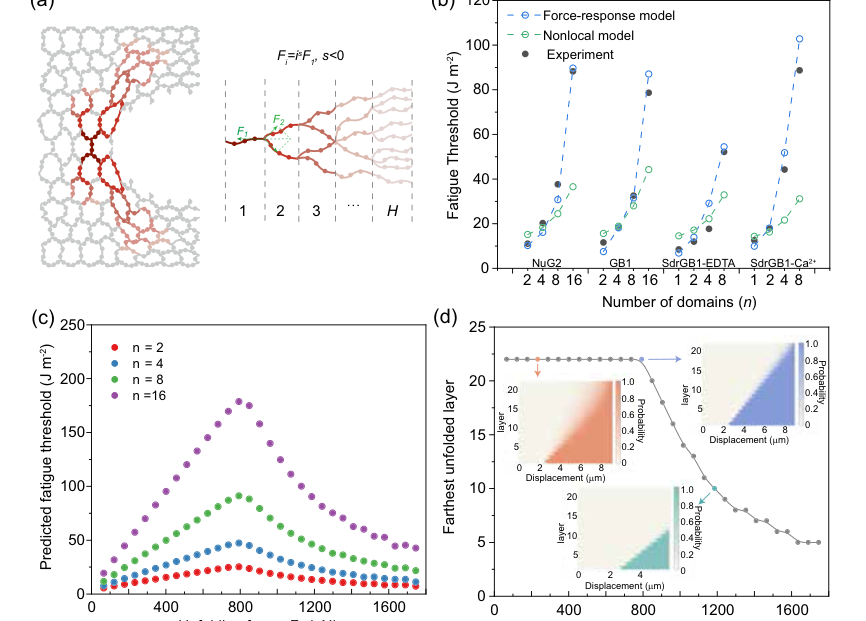 FIG. 5. Model prediction. (a) Schematic illustration of a fractured polymer chain along the crack path, deformed neighboring chains, and the force-response model with singular force decay across layers. (b) Comparison of fatigue thresholds among experiments, the prediction by the previous nonlocal model and the current force-response model of fabricated hydrogels with folded domains of GB1, NuG2, SdrGB1-EDTA, or SdrGB1-Ca2þ, when the number of folded domains varies within a single polymer chain. (c) The predicted fatigue threshold increases and then decreases with the unfolding force. (d) Predicted protein unfolding probability occurs away from the bridging strand for hydrogels with 16 domains in a polymer chain. The generation of farthest unfolded layers is governed by the force- dependent unfolding and folding kinetics of folded domains. For simplicity, we estimate the effect of unfolding force on the maximum extent of the unfolding zone by modifying the unfolding rate while fixing the folding rate (750/s) (see details in the Supplemental Material [25]). Below a threshold force, the unfolding zone can extend up to 23 layers, corresponding to a displacement of ∼8.5 μm. However, increasing the unfolding force beyond this threshold reduces the extent of the unfolding zone. Insets show the predicted displacement and the corresponding unfolding probability at distinct layers for three representative unfolding forces
FIG. 5. Model prediction. (a) Schematic illustration of a fractured polymer chain along the crack path, deformed neighboring chains, and the force-response model with singular force decay across layers. (b) Comparison of fatigue thresholds among experiments, the prediction by the previous nonlocal model and the current force-response model of fabricated hydrogels with folded domains of GB1, NuG2, SdrGB1-EDTA, or SdrGB1-Ca2þ, when the number of folded domains varies within a single polymer chain. (c) The predicted fatigue threshold increases and then decreases with the unfolding force. (d) Predicted protein unfolding probability occurs away from the bridging strand for hydrogels with 16 domains in a polymer chain. The generation of farthest unfolded layers is governed by the force- dependent unfolding and folding kinetics of folded domains. For simplicity, we estimate the effect of unfolding force on the maximum extent of the unfolding zone by modifying the unfolding rate while fixing the folding rate (750/s) (see details in the Supplemental Material [25]). Below a threshold force, the unfolding zone can extend up to 23 layers, corresponding to a displacement of ∼8.5 μm. However, increasing the unfolding force beyond this threshold reduces the extent of the unfolding zone. Insets show the predicted displacement and the corresponding unfolding probability at distinct layers for three representative unfolding forces
The first set of equations describes the singular force decay across hierarchical layers of the polymer network and the corresponding number of chains in each layer:
$$ F_i = F_1 \cdot i^{-s} \\ m_i = i^{-s} $$
The second set of equations, based on Bell's theory, quantifies the force-dependent unfolding and refolding kinetics of the protein domains in each layer:
$$ k_{uf,i} = k_{uf}^0 \exp\left(\frac{F_i z_{uf}}{kT}\right) \\ k_{f,i} = k_f^0 \exp\left(-\frac{F_i z_f}{kT}\right) $$
Term-by-Term Autopsy
Let's dissect each component of these master equations to understand their mathematical definitions, physical roles, and the rationale behind their formulation.
From the Force Decay and Chain Number Equations: $F_i = F_1 \cdot i^{-s}$ and $m_i = i^{-s}$
-
$F_i$:
- Mathematical Definition: The force acting on a single polymer chain in the $i$-th layer of the hierarchical network.
- Physical/Logical Role: This term quantifies the mechanical load experienced by the polymer strands at a specific "distance" or "generation" from the crack tip. It's crucial for understanding how stress is distributed away from the highly concentrated region near the crack.
- Why multiplication: The force in any given layer $i$ is expressed as a scaled version of the force in the first layer ($F_1$). Multiplication by $i^{-s}$ applies this power-law scaling factor to reflect the decay.
-
$F_1$:
- Mathematical Definition: The force acting on a single polymer chain in the first layer (i.e., the layer closest to the crack tip, where $i=1$).
- Physical/Logical Role: This represents the maximum or reference force experienced by the network, directly at the crack tip. It serves as the starting point for the singular force decay model.
- Why it's a term: It's the base value from which forces in all subsequent layers are derived, anchoring the force distribution to the crack-tip conditions.
-
$i$:
- Mathematical Definition: An integer index representing the $i$-th layer or generation of polymer chains, starting from $i=1$ for the layer closest to the crack tip.
- Physical/Logical Role: This variable denotes the hierarchical distance from the crack tip. It captures the spatial aspect of how forces and chain numbers change as one moves further into the bulk material.
- Why it's a term: It's the independent variable in the power-law relationship, indicating how the force and chain number vary with distance.
-
$s$:
- Mathematical Definition: A free parameter, referred to as the "singular exponent."
- Physical/Logical Role: This parameter governs the rate and shape of the force decay (and chain number distribution) with increasing layer number. A larger positive $s$ would imply a sharper, more localized force decay, while a smaller positive $s$ would mean a broader distribution. It reflects the network's topology and connectivity.
- Why it's an exponent: Power-law relationships, often characterized by singular exponents, are commonly used in fracture mechanics to describe stress fields near crack tips. This formulation provides a more realistic representation of stress concentration than simple exponential decay.
- Note on ambiguity: The paper states that $s$ "reflecting the spatial decay of force distributions" and then later mentions "the fitted singular exponent s is close to -1/2." If $s$ in the equation $F_i = F_1 \cdot i^{-s}$ is literally $-1/2$, then the term becomes $i^{-(-1/2)} = i^{1/2}$, implying that force increases with distance from the crack tip, which contradicts the concept of "decay." To be honest, I’m not completely sure about this part either, as it appears to be an inconsistency in the paper's description. For a physical "decay," the exponent of $i$ must be negative, meaning $s$ itself should be a positive value (e.g., if the exponent is $-1/2$, then $s=1/2$). I will proceed assuming the intended physical meaning of decay, where the exponent of $i$ is negative.
-
$m_i$:
- Mathematical Definition: The number of polymer chains in the $i$-th layer.
- Physical/Logical Role: This term represents the density or quantity of load-bearing polymer strands in a given layer. Its power-law distribution suggests that the effective number of chains contributing to the singular stress field also changes with distance from the crack tip.
- Why it's a term: It's a critical factor for calculating the total energy dissipated in each layer, as energy dissipation depends on both the force per chain and the number of chains.
-
$i^{-s}$:
- Mathematical Definition: A power-law scaling factor.
- Physical/Logical Role: This combined term dictates how both the force and the number of chains are scaled relative to the first layer, capturing the singular nature of stress distribution in fracture mechanics.
From the Kinetic Rate Equations (Bell's Law): $k_{uf,i} = k_{uf}^0 \exp\left(\frac{F_i z_{uf}}{kT}\right)$ and $k_{f,i} = k_f^0 \exp\left(-\frac{F_i z_f}{kT}\right)$
-
$k_{uf,i}$:
- Mathematical Definition: The unfolding rate of a protein domain in the $i$-th layer.
- Physical/Logical Role: This is the probability per unit time that a folded protein domain in layer $i$ will transition to an unfolded state under the influence of force $F_i$. It's a key parameter for quantifying the sacrificial energy dissipation.
- Why it's a term: It's a central kinetic parameter that determines how quickly the sacrificial bonds activate under mechanical load.
-
$k_{uf}^0$:
- Mathematical Definition: The intrinsic unfolding rate of a protein domain when no external force is applied ($F_i=0$).
- Physical/Logical Role: This represents the baseline rate at which a protein domain would unfold due to random thermal fluctuations alone. It sets the intrinsic kinetic scale for the unfolding process.
- Why it's a coefficient: It acts as a pre-exponential factor, scaling the force-dependent unfolding rate.
-
$\exp\left(\frac{F_i z_{uf}}{kT}\right)$:
- Mathematical Definition: An exponential factor derived from Bell's law.
- Physical/Logical Role: This term describes how the applied force $F_i$ exponentially accelerates the unfolding rate. Mechanistically, the force effectively lowers the energy barrier for unfolding, making the transition more probable.
- Why it's an exponential: Bell's law is rooted in transition state theory, where external forces bias the energy landscape, leading to an exponential change in reaction rates.
-
$z_{uf}$:
- Mathematical Definition: The unfolding transition distance.
- Physical/Logical Role: This molecular parameter represents the distance along the reaction coordinate from the folded state to the transition state for unfolding. It quantifies the sensitivity of the unfolding rate to force; a larger $z_{uf}$ means the rate is more strongly influenced by force.
- Why it's a term: It's a characteristic property of the protein domain, reflecting its mechanical stability and how it responds to external pulling forces.
-
$k$:
- Mathematical Definition: The Boltzmann constant.
- Physical/Logical Role: A fundamental physical constant that relates temperature to energy. It's essential for converting thermal energy into a comparable scale with mechanical work.
- Why it's a term: It's part of the thermal energy scale $kT$, which dictates the influence of thermal fluctuations on the kinetic processes.
-
$T$:
- Mathematical Definition: The absolute temperature of the system.
- Physical/Logical Role: Temperature provides the thermal energy available to the system. Higher temperatures generally increase the likelihood of overcoming energy barriers.
- Why it's a term: It's part of the thermal energy scale $kT$, directly influencing the kinetic rates.
-
$k_{f,i}$:
- Mathematical Definition: The refolding rate of a protein domain in the $i$-th layer.
- Physical/Logical Role: This is the probability per unit time that an unfolded protein domain in layer $i$ will refold. The negative sign in its exponent indicates that applied force typically hinders refolding, increasing the energy barrier. This reversibility is key to the hydrogel's fatigue resistance.
- Why it's a term: It's a crucial kinetic parameter that enables the "healing" or recovery aspect of the sacrificial bonds, allowing the network to dissipate energy repeatedly.
-
$k_f^0$:
- Mathematical Definition: The intrinsic refolding rate of a protein domain when no external force is applied ($F_i=0$).
- Physical/Logical Role: This represents the baseline rate at which an unfolded protein domain would refold due to thermal fluctuations alone. It sets the intrinsic kinetic scale for the refolding process.
- Why it's a coefficient: It acts as a pre-exponential factor, scaling the force-dependent refolding rate.
-
$\exp\left(-\frac{F_i z_f}{kT}\right)$:
- Mathematical Definition: An exponential factor with a negative exponent, derived from Bell's law.
- Physical/Logical Role: This term describes how the applied force $F_i$ exponentially decreases the refolding rate. The negative sign signifies that force typically increases the energy barrier for refolding, making the transition less probable.
- Why it's an exponential: Similar to unfolding, it's based on transition state theory, where force modifies the energy landscape.
-
$z_f$:
- Mathematical Definition: The refolding transition distance.
- Physical/Logical Role: This molecular parameter represents the distance along the reaction coordinate from the unfolded state to the transition state for refolding. It quantifies the sensitivity of the refolding rate to force.
- Why it's a term: It's a characteristic property of the protein domain, reflecting its refolding kinetics under force.
Step-by-Step Flow
Let's trace the journey of an abstract data point, representing the state of a single protein domain within a polymer chain in a specific layer $i$, as it passes through the mathematical engine during a simulated loading cycle. Imagine this as a mechanical assembly line:
- Global Stretch Input: The process begins with a prescribed global stretch applied to the entire hydrogel network. This is the initial "input" to our mechanical assembly.
- Force Distribution Calculation: This global stretch is then translated into local forces. The model first determines the force $F_1$ on the chains in the first layer (closest to the crack tip). Then, using the singular force decay law ($F_i = F_1 \cdot i^{-s}$), the forces $F_i$ on chains in all other layers $i$ are calculated. Simultaneously, the number of chains $m_i$ in each layer is also determined by $m_i = i^{-s}$. This step effectively distributes the overall load across the hierarchical network, creating a force profile.
- Kinetic Rate Evaluation: For each protein domain in each layer $i$, the calculated force $F_i$ is fed into the Bell's law equations.
- The unfolding rate $k_{uf,i}$ is computed: $k_{uf}^0$ (the intrinsic unfolding speed) is multiplied by an exponential factor that rapidly increases with $F_i$ (influenced by $z_{uf}$, $k$, and $T$). This tells us how likely the domain is to unfold under the current load.
- The refolding rate $k_{f,i}$ is also computed: $k_f^0$ (the intrinsic refolding speed) is multiplied by an exponential factor that rapidly decreases with $F_i$ (influenced by $z_f$, $k$, and $T$). This tells us how likely the domain is to refold.
These rates are like "switches" that determine the probability of state change.
- Domain State Update: Over a small, discrete time step, the model updates the state of the protein domains. If a domain is currently folded, it has a probability $k_{uf,i} \Delta t$ of unfolding. If it's unfolded, it has a probability $k_{f,i} \Delta t$ of refolding. This step advances the "fraction of unfolded domains" in each layer, reflecting the dynamic balance between unfolding and refolding.
- Network Mechanical Response Adjustment: When a protein domain unfolds, its effective contour length increases, making the polymer chain it belongs to longer and more compliant. Conversely, if it refolds, its contour length decreases. This change in the mechanical properties of the chains in layer $i$ is then fed back into the overall network mechanics, influencing how forces will be distributed in the next time step. This creates a feedback loop where molecular events impact macroscopic behavior.
- Energy Dissipation Accumulation: As the loading cycle progresses and forces are applied and removed, the unfolding and refolding events lead to a hysteresis in the force-extension curve for each layer. The area enclosed by this hysteresis loop for each layer $U_i$ represents the mechanical energy dissipated by that layer during the cycle.
- Total Energy and Fatigue Threshold Output: Finally, the energy dissipated in all layers is summed up to get the total energy $U$ consumed by the entire network. This total energy is then used in conjunction with other material properties to calculate the macroscopic fatigue threshold $\Gamma$, which is the ultimate output of the model, predicting the material's resistance to fatigue fracture.
This entire sequence is repeated iteratively for each small time step throughout the loading cycle, capturing the dynamic and history-dependent behavoir of the hydrogel under stress.
Optimization Dynamics
This theoretical framework is a simulation model rather than a machine learning algorithm, so it doesn't "learn" or "optimize" in the sense of gradient descent on a loss function. However, it does involve iterative updates and parameter calibration to match experimental observations.
-
Iterative State Updates and Convergence: The primary "update" mechanism within the model is the explicit time integration of the unfolding and refolding kinetics. At each small time step, the fraction of folded/unfolded domains in each layer is updated based on the current forces and kinetic rates. This iterative process allows the model to capture the dynamic, nonequilibrium, and history-dependent nature of energy dissipation. The system "converges" when the simulation reaches a steady state under cyclic loading, meaning the fraction of unfolded domains and the energy dissipated per cycle stabilize. This convergence represents the physical equilibrium (or pseudo-equilibrium under cyclic loading) of the system.
-
Parameter Calibration and Fitting: While the model itself doesn't autonomously "learn," its predictive power relies on carefully chosen or "optimized" parameters.
- Molecular Parameters: Parameters like the intrinsic unfolding/refolding rates ($k_{uf}^0, k_f^0$) and transition distances ($z_{uf}, z_f$) are typically derived from independent single-molecule force spectroscopy experiments or literature, providing a strong physical basis.
- Network Parameters: The singular exponent $s$ (from $F_i = F_1 \cdot i^{-s}$) is a free parameter that is fitted to experimental data. The paper mentions "the fitted singular exponent s is close to -1/2," indicating that an external optimization process (e.g., least-squares fitting) was used to find the value of $s$ that best reproduces the observed fatigue thresholds or other macroscopic mechanical properties. This fitting process is an external "optimization" where the model's predictions are compared against experimental results, and parameters are adjusted to minimize the discrepancy.
-
Implicit "Loss Landscape": Although not explicitly defined, one can conceptualize an implicit "loss landscape" where the "error" or "discrepancy" between the model's predicted fatigue thresholds (or other outputs) and the experimental data is the "loss function." The "optimization" then involves navigating this landscape (often through trial-and-error or more sophisticated fitting algorithms) to find the set of parameters that yield the best agreement. The "shape" of this landscape would indicate the sensitivity of the model's predictions to changes in its parameters. For instance, a steep landscape for $s$ would mean that small changes in $s$ lead to large changes in predicted fatigue thresholds.
In essence, the model's "learning" is not an internal, adaptive process but rather an external calibration of its physical parameters against experimental observations, allowing it to accurately predict the complex fatigue behavoir of hydrogels. The iterative nature of the simulation ensures that the dynamic molecular kinetics are fully accounted for in the macroscopic predictions.
Results, Limitations & Conclusion
Experimental Design & Baselines
To rigorously validate their claims, the authors meticulously engineered a series of hydrogels and subjected them to a comprehensive suite of mechanical and spectroscopic tests. The core of their experimental design revolved around incorporating custom-designed polyprotein crosslinkers, featuring folded protein domains, into a polyacrylamide network. These protein domains—NuG2, GB1, and SdrGB1—were chosen for their distinct and tunable mechanical stabilities. Specifically, SdrGB1's unfolding force could be modulated by the presence or absence of calcium ions, offering an elegant way to vary crosslinker strength without altering network topology.
The "victims" in this experimental setup were not just traditional models, but also the various hydrogel compositions themselves, allowing for direct comparison of how different protein domains and their numbers impacted fatigue resistance. The authors prepared 16 distinct hydrogel variants, all sharing identical polymer network structures (same polymer molecular weight and crosslink density) but differing in the type of protein crosslinker and the number of protein domains per crosslink (ranging from 1 to 16).
The experimental validation proceeded in several key steps:
1. Single-Molecule Force Spectroscopy (SMFS): Using an AFM-based SMFS system, the researchers directly measured the unfolding forces of individual protein domains. This step was crucial to confirm that the folded proteins indeed acted as "mechanically well-defined sacrificial units" with predictable force responses. They used eight tandem repeats of each protein domain to ensure robust measurements.
2. Bulk Mechanical Characterization: Tensile tests were performed on both unnotched and notched hydrogel samples. These tests assessed fundamental mechanical properties like Young's modulus, fracture stress, and strain, providing a macroscopic view of the hydrogels' behavior under load.
3. Fatigue Fracture Tests: Notched hydrogel samples were subjected to cyclic loading at a fixed frequency of 1 Hz. The primary metric for fatigue resistance was the fatigue threshold, defined as the maximum energy release rate (or strain energy intensity) at which a crack either does not grow or grows negligibly slowly per cycle. This direct measurement provided hard evidence of the hydrogels' durability under repeated stress.
The experimental architecture was designed to ruthlessly prove that the molecular-scale unfolding and refolding of protein domains directly influenced bulk fatigue behavior, offering a new paradigm for energy dissipation. The baselines for comparison included the traditional Lake-Thomas model and a previous nonlocal model, against which the new force-response model's predictions were benchmarked.
What the Evidence Proves
The evidence presented in this paper definitively demonstrates the efficacy of using folded protein domains as reversible, mechanically defined sacrificial units to enhance hydrogel fatigue resistance.
First, the single-molecule force spectroscopy (SMFS) results (Fig. 2) provided undeniable proof of the tunable mechanical properties of the protein crosslinkers. The characteristic sawtooth-like force-extension curves clearly showed individual unfolding events for each domain. The measured unfolding forces were distinct and consistent: NuG2 at approximately $112.6 \pm 47.2 \text{ pN}$, GB1 at $193.3 \pm 64.8 \text{ pN}$, SdrGB1-EDTA at $474.6 \pm 131.6 \text{ pN}$, and SdrGB1-Ca$^{2+}$ at a significantly higher $1492.7 \pm 130.5 \text{ pN}$. This directly validated the premise that these proteins serve as precise molecular force gauges, capable of dissipating mechanical energy at predetermined force thresholds.
Second, bulk mechanical tests (Fig. 3) revealed a crucial insight: despite varying protein domain types and numbers, all 16 hydrogel variants exhibited a similar Young's modulus of approximately $9 \text{ kPa}$. This is definitive evidence that the strategy enhances fatigue resistance without significantly increasing stiffness, a common and undesirable trade-off in many toughening strategies. While fracture stress and strain generally increased with the number of protein domains, their values did not simply scale with the unfolding forces, hinting at a more complex, distributed energy dissipation mechanism.
The most compelling evidence came from the fatigue fracture tests (Fig. 4). The fatigue thresholds consistently increased with the number of protein domains ($n$) for all hydrogel types. More strikingly, when comparing crosslinkers of equal length, the fatigue thresholds showed a nonlinear relationship with the unfolding forces of the protein domains. This counterintuitive finding—that networks incorporating weaker protein domains can achieve unexpectedly high fatigue thresholds—was a critical piece of evidence. It strongly supported the authors' hypothesis of "distributed energy dissipation," where a broader range of sacrificial events, rather than just high-force ruptures, contributes to overall toughness. This mechanism was shown to be highly localized near the crack tip, preventing extensive damage throughout the bulk material.
Finally, the theoretical force-response model (Fig. 5) provided a quantitative framework that beautifully aligned with the experimental observations. The model's predictions for fatigue thresholds showed a significantly better match to experimental data compared to the traditional Lake-Thomas and previous nonlocal models (Fig. 5b). This was the definitive, undeniable evidence that their core mechanism—coupling force redistribution with the kinetic unfolding and refolding of folded domains—accurately captures the real-world behavior of these hydrogels. The model further predicted an optimal unfolding force ($F_m$) for peak fatigue threshold (Fig. 5c), where domains are activated efficiently without premature exhaustion or remaining inactive. It also illustrated how the unfolding zone extends significantly for lower unfolding forces, demonstrating the distributed energy dissipation in action (Fig. 5d). This robust agreement between theory and experiment provides a powerful predictive tool for designing next-generation materials.
Limitations & Future Directions
While this work presents a brilliant and robust framework for understanding and designing fatigue-resistant hydrogels, it's important to acknowledge certain limitations and consider avenues for future development.
One inherent limitation lies in the simplifications of the theoretical model. Although significantly advanced, the force-response model still employs a hierarchical Cayley tree structure and, in its initial stages, assumed homogeneous chain behavior and uniformly folded domains. While the authors extended the model to resolve some of these, further refinement could incorporate more realistic network topologies, chain heterogeneity, and a wider distribution of domain unfolding forces to better capture the full complexity of real hydrogel networks. Additionally, the fitted singular exponent $s$ being close to $-1/2$ is a tentative finding, suggesting that further experimental and theoretical work is needed to fully characterize the stress-strain relationship in these complex materials.
Another practical limitation mentioned is that extremely weak proteins may unfold during gel swelling, which could complicate data interpretation and limit the range of usable protein domains. This suggests a need for careful selection and engineering of protein domains to ensure their stability under various environmental conditions, not just mechanical stress.
Looking ahead, this research opens several exciting discussion topics and future directions:
- Generalizing the Force-Response Framework: The authors suggest their framework can be extended to other classes of sacrificial interactions, such as hydrophobic associations or dynamic metal-ligand coordination, provided their force-dependent kinetics are known. A compelling future direction would be to apply this model to a broader range of soft materials incorporating different types of reversible sacrificial bonds, thereby establishing a more universal design principle for fatigue resistance.
- Precision Engineering for Optimal Performance: The model predicts an optimal unfolding force ($F_m$) for maximum fatigue threshold. Future research could focus on precisely engineering protein domains (or other sacrificial units) to hit this sweet spot, balancing the need for sufficient energy dissipation with preventing premature exhaustion or inactivity. This would involve a deeper understanding of how protein sequence and structure dictate unfolding kinetics and force.
- Exploring Multiscale Dynamics and History Dependence: The model explicitly integrates unfolding and refolding kinetics using Bell's law, capturing the history-dependent nature of energy dissipation. Future studies could delve deeper into how loading history, frequency, and temperature variations influence the long-term performance and "healing" capabilities of these hydrogels, potentially leading to adaptive materials that can self-repair or adjust their properties in response to environmental cues.
- Beyond Homogeneous Networks: While the current work focuses on random networks, future investigations could explore the impact of designed network topologies, such as gradient or hierarchical arrangements of protein domains with varying unfolding forces. Could a spatially varying distribution of weak and strong domains further optimize energy dissipation and crack delocalization?
- Integration with Other Toughening Mechanisms: This approach effectively enhances fatigue resistance without increasing stiffness. Future work could explore combining this protein-based sacrificial unit strategy with other established toughening mechanisms (e.g., double networks, fiber reinforcement) to create hydrogels with even more exceptional and multi-functional properties, while still maintaining low stiffness and high extensibility.
- Real-World Application and Biocompatibility: Given the biological origin of proteins, these hydrogels hold promise for biomedical applications. Future research should address long-term biocompatibility, biodegradability, and performance under physiological conditions, including potential immune responses or protein degradation pathways. This would be a crucial step towards translating these findings into clinical or industrial use.
By addressing these points, we can further evolve these findings, moving towards a more comprehensive understanding and predictive design of advanced soft materials with unprecedented durability.
Connections to Other Fields
Mathematical Skeleton
The pure mathematical core of this work involves modeling force distribution in a hierarchical network using a singular decay law, coupled with force-dependent kinetic equations for individual sacrificial elements, to predict macroscopic energy dissipation and fatigue thresholds. This framework integrates continuum-level stress singularities with molecular-level statistical mechanics of bond kinetics.
Adjacent Research Areas
Fracture Mechanics of Heterogeneous Materials
The paper's singular force decay law, $F_i = F_1 \cdot i^s$ (Eq. 1), directly relates to the concept of stress singularity at crack tips in classical fracture mechanics. In linear elastic fracture mechanics, stress often scales with $r^{-1/2}$, where $r$ is the distance from the crack tip. The parameter $s$ in the model effectively captures this spatial decay of force, extending these well-established principels to a network context where energy dissipation is distributed through sacrificial bonds. This approach is analogous to how toughening mechanisms, such as crack bridging or phase transformations in composites, are modeled to account for energy dissipation within a process zone ahead of the crack.
(Gdoutos, E. E., 2020, Fracture Mechanics, Solid Mechanics and Its Applications, Springer, Cham).
Mechanobiology and Polymer Physics of Force-Activated Processes
The kinetic equations for protein unfolding and refolding, $k_{uf,i} = k_{uf}^0 \exp\left(\frac{F_i z_{uf}}{kT}\right)$ and $k_{f,i} = k_f^0 \exp\left(-\frac{F_i z_f}{kT}\right)$ (Eq. 2), are a direct application of Bell's law, a fundamental model in mechanobiology for describing force-dependent molecular bond rupture or conformational changes. This law is widely used to understand how biological systems respond to mechanical stimuli at the molecular level. In polymer physics, the concept of sacrificial bonds that dissipate energy to enhance material toughness is a significant area of research, particularly in the design of tough hydrogels and elastomers. The paper's findings on an optimal unfolding force and the role of "weaker" bonds in damage delocalization align with principles explored in other mechanophore-containing polymer systems.
(Bell, G. I., 1978, Science, 200, 618-627).
Network Science and Statistical Physics of Disordered Systems
The use of a hierarchical Cayley tree structure to represent the hydrogel network and analyze force distribution within it connects strongly to network science and the statistical physics of disordered systems. These fields frequently study how local properties of nodes or bonds influence the global response of a network. The paper's investigation into how the number and strength of sacrificial protein domains affect the overall fatigue threshold is a specific instance of studying network robustness against failure. The observation that distributed energy dissipation through weaker bonds can enhance fatigue resistance resonates with studies on how heterogeneity and specific network topologies can improve the resilience of complex systems against localized damage, preventing catastrophic failure.
(Lin, S., & Zhao, X., 2020, Physical Review E, 102, 052503).